SALUD
¿Se está adaptando el virus de la viruela del mono al ser humano?
La Organización Mundial de la Salud (OMS) sopesa clasificar el brote de viruela del mono en países en los que la enfermedad no es endémica como una emergencia de salud pública de importancia internacional (ESPII), como la covid-19.

La Organización Mundial de la Salud (OMS) sopesa clasificar el brote de viruela del mono en países en los que la enfermedad no es endémica como una emergencia de salud pública de importancia internacional (ESPII). Es el nivel de alerta mundial más elevado, una distinción que exhiben la covid-19 y la polio. ¿Los motivos? Desde el 6 de mayo hasta el 22 de junio de 2022 han sido confirmados más de 3300 casos de viruela del mono en al menos 42 países diferentes. La enfermedad es endémica en países de África occidental y central, y hasta ahora los casos confirmados fuera de estas regiones habían estado asociados a importaciones desde zonas endémicas. Ahora la situación es diferente, por lo que el brote es considerado inusual y preocupante.
Por esa razón, los esfuerzos internacionales se han centrado en secuenciar al virus causante del brote, para caracterizar al patógeno, identificar el origen, rastrear la distribución, analizar la diversidad genética, orientar el diagnóstico, dirigir la investigación, evaluar la dinámica viral y conocer la trayectoria evolutiva del patógeno, entre otras facetas.
Pertenece al clado 3, de baja letalidad
Los resultados obtenidos han confirmado que todas las cepas del brote actual se agrupan estrechamente y que pertenecen al clado 3 de virus de la viruela del simio. El clado 3, al igual que el clado 2, está incluido en el linaje anteriormente denominado “África occidental”. El virus de la viruela del simio de los clados 2 y 3 es notificado con mayor frecuencia en zonas que van desde el oeste de Camerún hasta Sierra Leona. Por lo general, tiene una tasa de letalidad inferior al 1 %. En contraste, los virus del clado 1, anteriormente designados como linaje “África central”, son más agresivos y alcanzan una tasa de letalidad superior al 10%.
Lo más leído
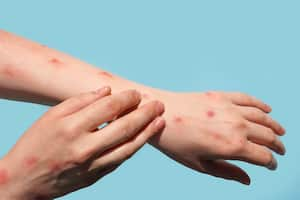
El rápido aumento de casos resulta inquietante. El agrupamiento sólido de las secuencias sugiere que el brote en curso tiene un único origen inicial. La transmisión silenciosa de persona a persona no parece factible, considerando las características conocidas de la enfermedad, que generalmente involucra lesiones cutáneas localizadas o generalizadas en las personas afectadas. Tampoco parece probable la transmisión críptica en un huésped animal en un país no endémico.
Sin embargo, cabe la posibilidad de que el desbordamiento veloz y la magnificación de la enfermedad haya ocurrido mediante eventos superpropagadores, cómo pueden ser reuniones sociales masivas con encuentros sexuales multitudinarios.
Está mutando a alta velocidad
Además, la situación actual nos lleva a un hecho preocupante, y es que los virus de la viruela del mono responsables del brote divergen una media de 50 SNP (Single Nucleotide Polymorphism) respecto a los virus de la viruela del mono más relacionados y que fueron responsables de los brotes ocurridos en los años 2018 y 2019 en los Estados Unidos, Israel y Singapur. Un SNP es una mutación puntual en la secuencia de ADN que afecta a un único nucleótido de una secuencia del genoma. Las estimaciones anteriores de la tasa de mutación en los orthopoxvirus, como es el virus de la viruela del mono, manejaban de 1 a 2 sustituciones por sitio por año, por lo que medio centenar de cambios en tan poco tiempo podrían representar una evolución acelerada del virus.
Muchas de las mutaciones encontradas hasta ahora son silenciosas porque no cambian ninguna de las proteínas virales. Sin embargo, al menos veintiuna de las mutaciones encontradas causan cambios en esas proteínas. En la actualidad es difícil predecir el efecto que pueden tener estas mutaciones individuales en el virus, pero en base a la función conocida de las proteínas virales de otros orthopoxvirus, y con el objetivo de ser analizadas y realizar un seguimiento exhaustivo, pueden ser clasificadas en prioridad baja, media y alta.

De estas mutaciones, hay tres (D209N, P722S, M1741I), que están localizadas en la proteína B21, que producen cambios en tres aminoácidos y que son clasificadas de alta prioridad. Estudios previos indican que las glicoproteínas de la familia B21/22 de la viruela del simio podrían ser un objetivo importante para los anticuerpos.
¿Un mutante que esquiva al sistema inmune?
Aunque las mutaciones del clado del brote están distribuidas por todo el genoma, es preocupante la existencia de un pequeño subconjunto de mutaciones en proteínas importantes que pueden estar involucradas en la transmisión del virus, la virulencia o la interacción con los medicamentos antivirales.

Por otra parte, la hipermutación observada puede sugerir la potencial acción de enzimas tipo APOBEC3, que son parte del sistema de defensa innato. Estas enzimas tienen la capacidad de editar el ADN vírico y bloquear la replicación del virus por lo que pueden inhibir a una amplia gama de virus mediante la introducción de mutaciones. Sin embargo, en algunas ocasiones, no logran interrumpir completamente la replicación del virus. Eso aumenta la probabilidad de que surjan variantes hipermutadas (“editadas” por las enzimas) pero viables, con características alteradas que les permiten, entre otras cosas, escapar a la respuesta inmune.

En este momento no se puede discernir si el exceso de mutaciones observadas en el virus de la viruela del simio es una consecuencia directa de la edición del genoma mediada por APOBEC3 en el huésped humano. Dado el escenario, la trayectoria actual de la incidencia de la enfermedad es incierta. Pero parece lógico pensar que han aparecido los primeros signos de microevolución de este virus durante la transmisión de persona a persona, por lo que es posible que esté en curso la adaptación del patógeno al ser humano.
Raúl Rivas González
Catedrático de Microbiología, Universidad de Salamanca
Artículo publicado originalmente en The Conversation