MUNDO
¿Qué se sabe de las mutaciones de covid-19 y cuál es el seguimiento científico?
La batalla contra la covid-19 no solo se libra en las salas de emergencia.
:quality(100)/cloudfront-us-east-1.images.arcpublishing.com/semana/XTQQ2FKNHBHADE3VNJLRV2UDR4.jpg)

Los científicos unen esfuerzos a nivel global en otra tarea monumental: descubrir cómo está mutando el virus que causa la enfermedad, el SARS-CoV-2. En enero, investigadores de China divulgaron la primera secuenciación del genoma del virus.
Desde entonces y hasta la fecha, más de 18.000 genomas del SARS-CoV-2, secuenciados por investigadores en diferentes países, se han depositado en una plataforma pública que permite compararlos y analizar sus diferencias.
Compartir abiertamente esa información es crucial. Permite saber no solo desde dónde y cuándo llegó el virus a un determinado lugar, sino ayudar a combatirlo.
En esta nota detallamos cómo se producen las mutaciones del SARS-CoV-2, cuál es la importancia de esos cambios y cómo los científicos los usan para rastrear el virus en ciudades como Nueva York y Montevideo.
Mutaciones frecuentes
"Una de las primeras cosas que debemos entender es que las mutaciones ocurren en todos los organismos", le explicó a BBC Mundo la científica Adriana Heguy, directora del Centro de Tecnología del Genoma de la Escuela de Medicina Grossman, de la Universidad de Nueva York, quien nació en Uruguay.

Crédito imagen: ADRIANA HEGUY dirige el Centro de Tecnología del Genoma de la Escuela de Medicina Grossman de la Universidad de Nueva York.
"A medida que las células de cualquier organismo se replican van adquiriendo mutaciones", explica. "En el caso de los virus ARN (cuyo material genético es ácido ribonucleico), como el que causa la covid-19, mutan mucho más rápido por el mecanismo intrínseco de la replicación, porque cuando el virus va haciendo copias de sí mismo la enzima que replica su genoma comete errores".
Al secuenciar el genoma de un virus ARN los científicos determinan el orden de los cuatro componentes básicos químicos, las llamadas "bases", simbolizadas por las letras A, G, C y U, que forman la molécula del ARN.
Y al comparar las diferentes secuencias es posible identificar mutaciones que, para hacerse una idea, podrían considerarse como errores tipográficos que ocurren durante el proceso de copia.
Sin embargo, la palabra mutación no necesariamente debe ser motivo de alarma.
Mutación no equivale a mayor riesgo
Heguy señala que "la mayor parte de las mutaciones probablemente no tienen ningún efecto, así como la mayor parte de las variaciones genómicas entre una persona y otra no tienen ningún efecto en materia de enfermedad".
Lo más leído
"Pero algunas de estas mutaciones sí pueden tener efecto sobre el virus", añade. Recientemente, varios medios informaron sobre un trabajo preliminar en torno al tema del Laboratorio Nacional de Los Álamos, en Estados Unidos, aún no revisado formalmente por pares (científicos independientes a la investigación).
Crédito imagen: GETTY IMAGES. No hay ninguna evidencia sólida en este momento para pensar que una mutación es más transmisible o causa síntomas más graves de covid-19, según Heguy.
El estudio indica que una mutación en una de las proteínas del virus lo haría más contagioso.
Sin embargo, varios expertos aseguran que aún no hay consenso sobre si una de las mutaciones del nuevo coronavirus es más peligrosa que otras.
En otras palabras, la hipótesis es posible en principio, pero la evidencia en el estudio no lo demostraría.
"No hay ninguna evidencia sólida en este momento para pensar que esa mutación o cualquier otra mutación es más transmisible o causa síntomas más graves de covid-19, aunque es válido seguir investigando este tema", le afirmó Heguy a BBC Mundo.
Muchas mutaciones, una cepa
Vincent Racaniello, profesor de microbiología de la Universidad de Columbia, advierte sobre el error en los medios de magnificar la importancia de cada mutación.
"Por el momento solo hay una cepa de SARS-CoV-2", asegura Racaniello en su blog virology.ws. "Cepa" es un término usado a veces en microbiología en forma más laxa como "variante".
Sin embargo, para Racaniello, debe reservarse estrictamente para variantes con propiedades biológicas diferentes y hasta ahora "nadie ha demostrado" que exista un virus aislado de algún paciente con esas características.

Crédito imagen: GETTY IMAGES. Varios expertos aseguran que aún no hay consenso sobre si hay una mutación de coronavirus más peligrosa que otras.
"Lo que justificaría hablar de una nueva cepa serían, por ejemplo, los cambios en la virulencia (la capacidad de causar enfermedad), en la habilidad del virus de ser bloqueado por una respuesta inmunológica o en la estabilidad del virus a altas temperaturas", le explicó Racaniello a BBC Mundo.
Hasta ahora, según el investigador, "los virus aislados de muestras de pacientes son diferentes en la secuencia de sus genomas, pero tienen las mismas propiedades biológicas, por lo que son la misma cepa".
Miles de genomas disponibles en internet
Los miles de genomas secuenciados están siendo depositados en una base de datos internacional de acceso abierto llamada Gisaid, que fue creada en 2008 para compartir datos sobre el virus de la gripe.
Gisaid es el acrónimo en inglés de Iniciativa Global para Compartir Datos sobre Influenza, o Global Initiative on Sharing All Influenza Data.
"Es un esfuerzo colaborativo mundial y prácticamente todo el mundo que está trabajando sobre el coronavirus está subiendo datos genómicos a esa base de datos", le afirmó a BBC Mundo el biólogo Gregorio Iraola.
Iraola es el responsable del Laboratorio de Genómica Microbiana del Institut Pasteur de Montevideo y ha contribuido hasta ahora al Gisaid con 10 genomas.

Crédito imagen: RICHARD NEHER. Richard Neher, profesor de la Universidad de Basilea, es uno de los fundadores de Nextstrain.
Otra plataforma clave en el estudio de la covid-19 es Nextstrain (se traduce como "próxima cepa"), un sitio fundado tras una conversación entre dos jóvenes investigadores que se encontraron en una conferencia en California en 2014.
"En ese momento queríamos crear un sitio que proveyera análisis siempre actualizados sobre la influenza", le explicó a BBC Mundo uno de sus fundadores, Richard Neher, investigador en evolución de virus y bacterias de la Universidad de Basilea, en Suiza.
Nextstrain recoge los miles de genomas del virus de covid-19 que se van subiendo a Gisaid y los analiza en tiempo real.
Con esa información elabora árboles de parentesco que muestran las relaciones entre los genomas del virus, lo que se conoce como "árboles filogenéticos".
Y conociendo los tiempos y los lugares en que las mutaciones fueron detectadas, Nexstrain elabora mapas que reconstruyen las migraciones del virus de un lugar a otro.
Cómo las mutaciones permiten rastrear el virus
Comparar las mutaciones gracias a plataformas como Gisaid y Nextstrain permite a los científicos rastrear cómo se esparce el virus en el mundo.
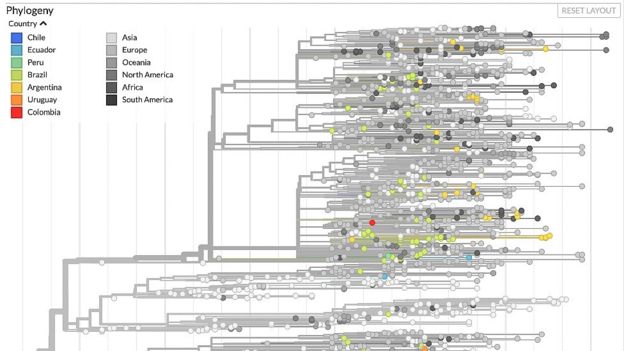
Crédito imagen: GENTILEZA NEXTSTRAIN. Las mutaciones permiten rastrear cómo se desplaza el virus en mapas como el de la plataforma Nextstrain.
"A través de esas mutaciones le podemos seguir la pista al virus, en la misma forma en que si yo hago tu genoma, y se lo hago a gente de tu familia, te puedo decir quién es tu hermano y saber que venís de tu mamá y de tu papá y no de los míos", afirmó Adriana Heguy.
"Lo que hacemos es comparar genomas y ver que tienen cambios genéticos, marcas que podemos leer y vendrían a representar una especie de código de barras específico de cada genoma", explicó Iraola.
Si las mutaciones del virus en algún sitio del planeta tienen grandes similitudes con las de genomas obtenidos con anterioridad en otra parte del mundo, esas semejanzas pueden indicar tras un complejo análisis estadístico que el virus "viajó" de un país a otro.
Ese es precisamente el análisis que realizaron los científicos en Nueva York y Montevideo.
De Europa a Nueva York
En el caso de Nueva York, dos estudios que aún aguardan revisión por pares llegaron a conclusiones similares a pesar de analizar muestras de pacientes diferentes.
Una de las investigaciones fue dirigida por Heguy y se basó en muestras de 236 afectados.
"Cuando nosotros secuenciamos las muestras de Nueva York, prácticamente en el 70 % descubrimos que se trata de cepas europeas", algo que resulta revelador.

Crédito imagen: GETTY IMAGES. Científicos descubrieron una conexión directa entre la cepa del virus europea y neoyorquina.
El presidente Donald Trump suspendió los vuelos desde Europa el 11 de marzo.
Antes de eso "no se controlaba a ninguna persona, ni se les decía que se quedaran en casa cuando volvían de Europa. Era evidente para los epidemiólogos que el virus no se iba a quedar tranquilito en China", afirmó Heguy.
"Yo pensé que íbamos a encontrar más variantes significativas chinas, pero se ve que interrumpir los vuelos de China y decirle a la gente que venía de allí que se quedara en casa por 14 días tuvo un impacto, porque nosotros encontramos pocas variantes que son directamente asiáticas".
Más evidencia de la conexión Europa-Nueva York
La autora principal del otro estudio sobre Nueva York es la científica guatemalteca Ana Silvia González Reiche, investigadora posdoctoral en el Departamento de Genética y Genómica de la Escuela de Medicina Icahn del Hospital Monte Sinaí en Nueva York.
Le contó a BBC Mundo que empezaron a secuenciar genomas la primera semana de marzo, que llevan hechas las secuencias de 400 y que siguen trabajando en ello.

Crédito imagen: GETTY IMAGES. "Analizando las relaciones genéticas entre los virus determinamos que la mayoría eran más cercanos y tenían descendientes en común con los virus europeos", afirmó la científica guatemalteca Ana Silvia González-Reiche.
"La mayoría de los casos estudiados provienen de introducciones no documentadas por rutas desconocidas y es aquí donde la información genética nos ayuda reconstruir el pasado de estas infecciones".
Tres rutas a Uruguay
En el caso de Uruguay, Iraola y sus colegas secuenciaron 10 genomas de muestras de pacientes durante la fase temprana de la epidemia en el país a mediados de marzo.
"Nosotros encontramos en estos 10 casos tres introducciones independientes a Uruguay de tres continentes distintos: una desde Europa, más concretamente desde España; otra desde Norteamérica, muy posiblemente desde Canadá, y la tercera desde Australia", afirmó el investigador del Institut Pasteur.
"Además, pudimos estimar la fechas más probables de esas introducciones, que nos indican que hay posibilidad de que hayan sido previas a los primeros casos reportados en el país ".

Crédito imagen: DANIELA HIRSCHFELD. Gregorio Iraola, del Institut Pasteur de Montevideo, comparó genomas de distintas partes del mundo para determinar las rutas de entrada del virus a Uruguay.
La comparación de genomas permite no solo identificar desde dónde viajó el virus, sino cuándo.
"Eso es posible porque las mutaciones se van acumulando en el genoma a un determinado ritmo, a una determinada tasa; es decir, ocurren cada determinada unidad de tiempo, lo que se llama reloj molecular", afirmó Iraola, cuyo estudio acaba de ser divulgado y aguarda revisión por pares.
"Nosotros sabemos que el virus se detectó en Uruguay el 13 de marzo, y tenemos datos de todos los virus secuenciados de todo el mundo de los cuales conocemos su país de origen y la fecha exacta en la que se detectaron".
"Es básicamente como un reloj. Comparando las tasas de cambio genético podemos estimar cuál fue la fecha en la que ese virus estuvo presente antes en otro lugar."
Preguntas sin respuesta
Heguy ha secuenciado genomas del virus de unos 500 pacientes y espera llegar a mil.
En cada uno de esos pacientes la investigadora y sus colegas monitorean parámetros clínicos como la severidad de la enfermedad o el tiempo que la persona permanece hospitalizada.
"Una de las cosas que estamos tratando de averiguar es si hay una correlación entre esos parámetros clínicos con ciertas mutaciones y tratar de entender el efecto que estas mutaciones del virus pueden tener sobre la enfermedad en sí".
Crédito imagen: SCIENCE PHOTO LIBRARY. Aún no se sabe cómo las mutaciones pueden impactar los esfuerzos para hallar una vacuna.
Otra pregunta que muchos se hacen es si las mutaciones afectarán los esfuerzos para hallar una vacuna.
"Esto aún no se sabe. Por eso es importante continuar secuenciando genomas para monitorear la evolución del virus a través del tiempo", señaló Ana Silvia González Reiche.
"No lo sabemos todavía, pero podría ser una posibilidad. Este virus es muy nuevo", afirmó por su parte Adriana Heguy.